OVERVIEW
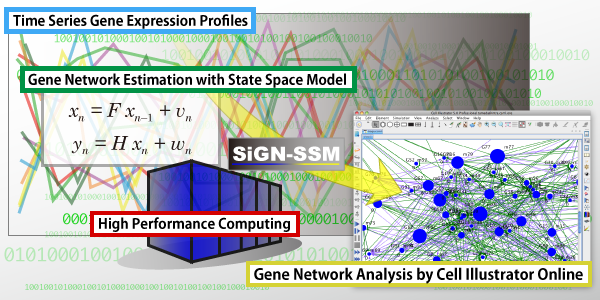
SiGN-SSM is open source gene network estimation software able to run in parallel on PCs and massively parallel supercomputers. The software estimates a state space model (SSM), that is a statistical dynamic model suitable for analyzing short time and/or replicated time series gene expression profiles. SiGN-SSM implements a novel parameter constraint effective to stabilize the estimated models. Also, by using a supercomputer, it is able to determine the gene network structure by the statistical permutation test in a practical time. SiGN-SSM is applicable not only to analyzing temporal regulatory dependencies between genes, but also to extracting the differentially regulated genes from time series expression profiles.
SiGN-SSM is distributed under GNU AFFERO GENERAL PUBLIC LICENCE (GNU AGPL) version 3. The pre-compile binaries for Linux (x86-64), MS Windows, and Mac OS X are also available in addition to the source code. The pre-installed binaries are available on the Human Genome Center (HGC) supercomputer system [6] and R-CCS K computer [7].
NEWS
FEATURES
CONTENTS
How to use SiGN-SSM : Online tutorial of SiGN-SSM.
Manual : User reference manual.
Download : Source code and pre-compiled binary executable.
Contact : Contact information & developer list
PUBLICATION
The main paper describing SiGN-SSM is Tamada et al. (2011) [1] in REFERENCE below. Please cite this if you want to refer to SiGN-SSM in your publication, etc. The mathematical details are described in Hirose et al. (2008) [2] and Yamaguchi et al. (2008) [3].
The example of the application to the real data analysis is Yamauchi et al. (2012) [8].
ACKNOWLEDGEMENTS
SiGN-SSM is developed in the ISLiM (Next-generation integrated simulation of living matter) project in RIKEN Computational Science Research Program [5]. This is based on the previous implementation TRANS-MNET [2]. Computational resources required for the development of SiGN-SSM was provided by the HGC Supercomputer System, Human Genome Center, Institute of Medical Science, The University of Tokyo; and RIKEN Supercomputer system RICC.
REFERENCE
[1] Tamada, Y., Yamaguchi, R., Imoto, S., Hirose, O., Yoshida, R., Nagasaki, M., and Miyano, S. (2011). SiGN-SSM: open source parallel software for estimating gene networks with state space models. Bioinformatics 27 (8), 1172-1173.
[2] Hirose, O., Yoshida, R., Imoto, S., Yamaguchi, R., Higuchi, T., Charnock-Jones, D.S., Print, C., and Miyano, S. (2008). Statistical inference of transcriptional module-based gene enetworks from time course gene expression profiles by using state space models. Bioinformatics 24 (7), 932-942.
[3] Yamaguchi, R., Imoto, S., Yamauchi, M., Nagasaki, M., Yoshida, R., Shimamura, T., Hatanaka, Y., Ueno, K., Higuchi T., Gotoh, N., and Miyano, S. (2008). Predicting differences in gene regulatory systems by state space models. Genome Informatics 21, 101-113.
[4] Wu, L.S.-Y., Pai, J.S, and Hosking J.R.M. (1996). An algorithm for estimating parameters of state-space models. Statistics & Probability Letters 28, 99-106.
[5] LINK: RIKEN Computational Science Research Program
[6] LINK: HGC Supercomputer System
[7] LINK: RIKEN AICS "K computer"
[8] Yamauchi, M., Yamaguchi, R., Nakata, A., Kohno, T., Nagasaki, M., Shimamura, T., Imoto, S., Saito, A., Ueno, K., Hatanaka, Y., Yoshida, R., Higuchi, T., Nomura, M., Beer, D.G., Yokota, J., Miyano, S., and Gotoh, N. (2012). Epidermal growth factor receptor tyrosine kinase defines critical prognostic genes of stage I lung adenocarcinoma. PLoS ONE 7(9), e43923. (See at PLoS ONE)